

男性,17岁
发现右侧膝关节旁包块就诊
病案讨论
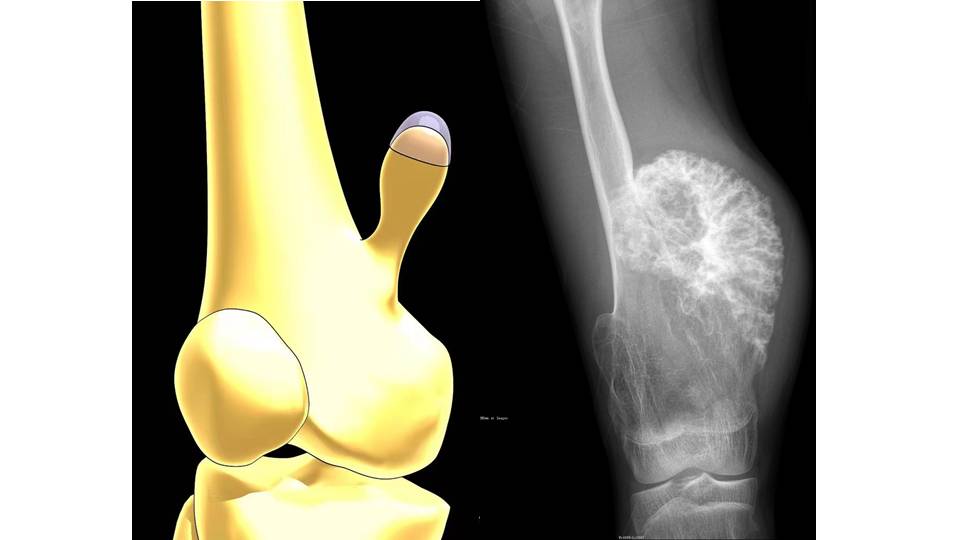
影像表现:右侧股骨近端及远端见多发骨性突起,背离关节,其中右侧内髁上方旁骨性突起较大,表面边缘密度增高钙化改变,同时左侧胫腓骨近端及远端均见多发骨性突起,背离关节,皮质与髓腔均与母体骨相延续,其中左侧胫骨下端外侧突起明显压迫腓骨,使其骨质吸收变细弯曲。影像诊断为典型的多发骨软骨瘤。
最终诊断:遗传性多发性骨软骨瘤病。
遗传性多发骨软骨瘤病(Hereditary Multiple Exostoses, HME),又称遗传性多发外生骨疣、骨干连续症等,是一种较少见的常染色体显性遗传性疾病,男女比例约3∶1,以四肢长骨多发,多见于股骨、胫骨远近端、尺桡骨远端及肱骨近端等部位。病因尚未完全明确,可能与先天性胚胎缺陷、骨骺板错置、骨骼干骺端在骨骼生长过程中失去塑形能力导致干骺端增宽并增殖,以及由骨膜内层残存的幼稚细胞或化生成的软骨细胞逐渐生长等原因导致。研究认为此类疾病为EXT1、EXT2基因突变导致,而目前报道的EXT1基因突变超过200个,EXT2基因突变有近100个,最新研究报道了EXT2的2处新突变位点,约90% 的多发性骨软骨瘤为显性遗传。临床上表现为膝关节周围多发质硬包块,前臂短小、弯曲,腕、肘关节显著畸形和活动障碍,从而严重影响到患肢的外观和功能。
病理组织学表现: ①表层为血管稀少的胶原结缔组织与周围骨膜衔接,并紧密附着于其下方组织; ②中层为灰蓝色透明软骨,即“软骨帽”,类似正常软骨,常发生钙化; ③基层为肿瘤主体,含黄骨髓的骨松质与患骨相连。
影像表现:干骺端增粗变宽,骨皮质变薄,骨赘向旁侧突起,背向关节,骨疣呈宽基底或蒂状、菜花状,肿块较大时致相应骨干膨大、畸形,部分骨疣顶部或周围可见散在钙化斑。多发性骨软骨瘤病患者中90% 病变是宽基底的,只有10% 的骨疣有蒂。骨软骨瘤恶变的影像学表现是: 软骨帽持续增厚,帽四周非均匀钙化,出现透亮区,大的含软骨基质的软组织肿块,蒂遭到侵蚀破坏,瘤体较前明显增大; 基底部及骨干骨皮质溶骨性破坏; 周围软组织明显肿胀。本例为股骨、胫腓上下端多发的骨软骨瘤,宽基底,符合HME诊断,其中右侧股骨下端骨软骨瘤瘤体较大,但表面钙化且边缘较光滑,未见明显钙化影吸收破坏,未见明显软组织肿块,邻近皮肤软组织均为肿瘤压迫改变,本例行手术切除后病理显示为单纯骨软骨瘤,并非肿瘤恶变。
治疗:大多数的骨软骨瘤患者是无症状的且不需要治疗。多主张出现局部疼痛妨碍关节活动或压迫血管、神经和脏器时,才是手术切除的指征。骨软骨瘤的切除术应整块切除,肿瘤的蒂或基底部周围应有一圈正常的骨组织,还要包括软骨帽、骨膜和整个滑囊一并切除。如果成年后肿瘤仍在生长,要引起高度重视,可能是发生恶变的征兆。研究发现早期手术单纯切除骨软骨瘤,并不能避免骨骼畸形的发生,也不能增加骨软骨瘤的癌变的发生率。多发性骨软骨瘤的恶变率较高,多为软骨肉瘤。其发病率为0.5% ~ 2%但软骨肉瘤恶性程度较低,早期手术切除后预后较好。 Bovee随访一例恶变为软骨肉瘤者 12 年,手术切除治疗未复发。当患处疼痛加重或者骨骼发育成熟后仍有生长应考虑骨软骨瘤发生恶变,大多数的骨软骨瘤患者是无症状的且不需要治疗。预防性切除无症状病变并不能防止骨软瘤恶变。多发性骨软骨瘤的治疗尚未取得突破性进展,本病最根本的治疗可能是寻找遗传基因,进行基因治疗。
类型:原创
病例ID:ZYLM000003681
校对:王宇军
阅读:1562
文章已于2023-11-21修改




